



(1) Overview
Introduction
Statistical analysis of data on twins focuses on determining the relative contributions of nature and nurture to individual differences in a behavioral trait (i.e., phenotype). Twin pairs are either identical (monozygotic, MZ) and share the same genomic sequence or non-identical (dizygotic, DZ) and share on average only half of the segregating genes. When MZ twin pairs are more similar in a phenotype (e.g., depression or educational achievement) than DZ twin pairs, this implies that genetic influences are important.
Traditionally, in the classical twin study, the ACE model is used which decomposes total phenotypic variance (e.g., the total variance in the scores on a depression scale or mathematics test), 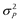 , into variance due to additive genetic (A) influences (
, into variance due to additive genetic (A) influences ( ), common-environmental (C) influences (
), common-environmental (C) influences ( ) and unique-environmental (E) influences (
) and unique-environmental (E) influences ( , residual variance) []. Common-environmental influences are shared influences that lead to alikeness between twins that cannot be attributed to their genetic resemblance and are parametrized as being perfectly correlated within one twin pair. Unique-environmental influences are not shared in the family and are parametrized as being uncorrelated for members of a twin pair. It is also possible to fit an ADE or AE model. In the ADE model, the C component is replaced by a D component (representing dominance effects, that is, non-additive genetic influences) and in the AE model the C component is fixed to 0. Furthermore, an interaction between genetic- and environmental influences (G × E) can be added to the model in which, conditionally on the genotypic value of a twin, unique-environmental influences can be either more or less important. Usually, for the collection of phenotypic data, a questionnaire or test is used where respondents answer a set of items, assuming that they are representative for the underlying trait of interest.
, residual variance) []. Common-environmental influences are shared influences that lead to alikeness between twins that cannot be attributed to their genetic resemblance and are parametrized as being perfectly correlated within one twin pair. Unique-environmental influences are not shared in the family and are parametrized as being uncorrelated for members of a twin pair. It is also possible to fit an ADE or AE model. In the ADE model, the C component is replaced by a D component (representing dominance effects, that is, non-additive genetic influences) and in the AE model the C component is fixed to 0. Furthermore, an interaction between genetic- and environmental influences (G × E) can be added to the model in which, conditionally on the genotypic value of a twin, unique-environmental influences can be either more or less important. Usually, for the collection of phenotypic data, a questionnaire or test is used where respondents answer a set of items, assuming that they are representative for the underlying trait of interest.
Item Response Theory
Commonly, when twin data are analysed, the phenotypic data of an individual twin is aggregated to a single score for example by calculating a twin’s sum over all answered items (i.e., the sum score). However, earlier research has shown that using an aggregated score such as the sum score can lead to an underestimation of heritability [] and the spurious finding of G × E effects as artefact of scale properties such as heterogeneous measurement error [, ].
Earlier research has also shown that the incorporation of an item response theory (IRT) measurement model into the genetic analysis can overcome this potential bias [, , ]. The IRT approach is model-based measurement in which a twin’s latent trait (e.g., mathematical ability) is estimated using not only trait levels (e.g., performance on a mathematics test) but also test item properties such as the difficulty or discrimination of each item are taken into account. The simplest IRT model is the one-parameter logistic model (1PLM), which models the probability of a correct answer to item k (e.g., of a mathematics test) by twin j from family i, P(Yijk = 1), as a logistic function of the difference between the twin’s latent trait score (e.g. mathematical ability) and the difficulty of the item:

where the difficulty parameter of item k, βk is parametrized as the trait level (e.g., the mathematical ability level) needed to have a 50% chance of answering item k right (or, in case of a personality test, responding “yes” to an item). While this model assumes that all items discriminate equally well between varying traits, in the two-parameter model (2PL), an extension of the 1PL model, also discrimination parameters (comparable to factor loadings) that differ between items are estimated [].
The 1PL and 2PL are suitable for dichotomous data (e.g., scored as correct = 1 and false = 0), as for example collected from ability tests (e.g., mathematical ability). For non-dichotomous data such as ordered categories (e.g., Likert scale data) as often collected in personality tests, the partial credit model (PCM, without different discrimination parameters) or the generalized partial credit model (GPCM, with different discrimination parameters) can be used []. The PCM (GPCM) can be considered as an extension of the 1PL (2PL) that treats polytomous responses as ordered performance levels, assuming that the probability of selecting the kth cateogry over the [k – 1] category is governed by the dichotomous (1PL or 2PL respectively) IRT model [].
Besides the ones above mentioned, further advantages of the IRT approach include the flexible handling of missing data and the harmonization of traits measured on different measurement scales. For example, when different twin registers have used different IQ tests not comparable in difficulty, IRT can be used to set the items scores on the same measurement scale [].
Simultaneous estimation through MCMC algorithms
While free and open-source software for analysis of twin data is available [], the software restricts the user to perform an item-level analysis using a two-step approach. That is, in the first step, an IRT model is analysed and in the second step, the resulting latent traits are used in a variance decomposition. This approach, however, does not solve the psychometric issues outlined in earlier research [, , ]. For example, measurement erorr is included in the estimated latent traits and the two-step approach cannot correct for bias through ceiling effects when the 1 PL model is applied [].
To take full advantage of the IRT approach, both genetic and IRT model have to be estimated simultaneously [], which requires the evaluation of multiple integrals when the tradional frequentist approach is used for inference. We can avoid this problem by adopting a Bayesian approach and using Markov chain Monte Carlo (MCMC) estimation procedures. In Bayesian analysis, statistical inference is based on the joint posterior density of the model parameters, which is proportional to the product of the likelihood function and a prior probability distribution. A prior probability distribution represents information about an uncertain parameter before any data has been observed.
Although MCMC modelling of twin data is easily carried out using off-the-shelf software packages like JAGS [] and BUGS [] and example syntax is published in journals available to the twin community [, , , , ], the twin research community seems reluctant to embrace the new technology with its richness of possibilities for two reasons: First, most researchers in the field of behavior genetics have an applied background and are therefore less familiar in learning a new programming language and/or adopting the available syntax to their own needs. Second, the use of Bayesian statistics is not that common yet in the field of behavior genetics which makes it more difficult for behavior geneticists to decide what information is relevant and how the output should be interpreted.
To facilitate the use of Bayesian statistics and prevent bias in heritability and/or the spurious finding of G × E interactions by analysing both IRT and genetic model simultaneously, the R package BayesTwin serves as a pipeline from R to the open-source MCMC software program JAGS []. The package includes all common univariate genetic twin models (ACE, AE, ADE) and can analyse dichotomous (using a 1PL or 2PL IRT model) as well as polytomous item-level data (using a PCM or GPCM). Optionally, G × E can be estimated and/or covariates can be included in the model. G × E is parametrized such that both genetic as well as environmental, influences are modelled as latent (i.e., unmeasured) variables, representing a powerful omnibus test to assess whether there is any statistically significant interaction. An interaction is considered on unique-environmental influences where is portioned into an intercept (i.e., representing average environmental variance) and a part that is a function of the genotypic value (i.e., representing G × E). For more technical detail, the reader is referred to earlier publications that used this G × E parametrization [, , , ]. Covariates are integrated into the model such that the phenotypic variance decomposition takes place after the effects of the covariates have been partialled out, but other than that the covariate data are not part of the covariance model [].
Implementation and architecture
The main function IRTtwin () can be used to analyse item-level twin data. Furthermore, a function is included that can be used to simulate item-level item data (simulatetwin ()). In addition, BayesTwin includes functions that determine whether the Bayesian analyses was performed well or help plot relevant information in figures and compute posterior statistics such as posterior mean, posterior standard deviations, and 95% highest posterior density (HPD) intervals.
A requirement to use the main function IRTtwin is that the data of MZ and DZ twins (both phenotypic as well as covariate data) need to be stored in two different matrices, one including the data of all MZ twin pairs and one including the data of all DZ twin pairs (function arguments data_mz and data_dz). The function then requires as input the specific data columns in which the phenotypic and covariate data of the first and second twin is stored (twin1_datacols_p, twin2_datacols_p, twin1_datacols_cov and twin2_datacols_cov). Furthermore, the genetic and IRT model to analyse the data has to be specified (decomp_model and irt_model), whether a G × E interaction effect should be included (ge = TRUE), the number of covariates (N_cov) and the number of categories when ordinal data is used (Nk) and whether fit statistics should be calculated (fit_stats = TRUE). Optional additional arguments concern technical details (e.g., the number of iterations for the MCMC algorithm, the number of Markov chains, the choice of initial values and the choice of the prior distribution for variance components). Below, you can find the R code that analyses data on 40 dichotomous items under the ACE with G × E, including 2 covariates and using the 1PL IRT model:
IRTtwin (data_mz = data$y_mz, data_dz = data$y_dz,
twin1_datacols_p = 1:20,
twin2_datacols_p = 21:40,
twin1_datacols_cov = 41:42,
twin2_datacols_cov = 43:44,
decomp_model = “ACE”, irt_model = “1PL”,
ge = TRUE,
N_cov = 2, inits = NA, Nk = 0,
fit_stats = FALSE,
n_iter = 10000, n_burnin = 8000,
n_chains = 1,
var_prior = “INV_GAMMA”, inits = NA)
The retrieved output includes posterior samples, posterior point estimates with standard deviations and the 95% HPD interval for variance components and, if applicable, regression parameters. The HPD can be seen as the Bayesian version of a confidence interval. All objects that are returned from the main function are automatically assigned the class “bayestwin”. The S3 method summary.bayestwin () can then be used to get a nicely formatted table in R that includes summary statistics for variance components, and, if applicable, regression parameters.
The function plotbayestwin () can be used to plot posterior distributions and trace lines. For an example of both plot types see Figure 1. When a model is estimated that includes G × E, the function geplot () can be used to plot the 95% credibility region of the interaction effect (see Figure 2).
Example of a trace plot (a) and posterior density plot (b) produced by the function plotbayestwin ().
Example of a G × E interaction plot produced by the function geplot ().
Quality control
The code is based on earlier publications [, , ] that used simulation studies to test the newly developed methodology. Furthermore, multiple smaller and large code review were held throughout the development process. The structure of the package successfully passed the CRAN R CMD check. Results from this check can be found on CRAN (https://cran.r-project.org/web/packages/BayesTwin/index.html).
(2) Availability
Operating system
Since BayesTwin is written in R and uses JAGS for the MCMC sampling algorithm, it should run on any system on which R, JAGS and the BayesTwin dependencies run. R and JAGS can be obtained freely from https://www.r-project.org/ and http://mcmc-jags.sourceforge.net/. BayesTwin was tested on Apple Mac OS X 10.9 and Windows 7.
Programming language
R version 3.4.0 or higher and JAGS version 4.2.0 or higher.
Additional system requirements
An Internet connection is required to install the BayesTwin package and JAGS.
Dependencies
R packages: foreign, coda, matrixStats and rjags.
List of contributors
The package was created by Inga Schwabe.
Software location
Archive
Name: CRAN
Persistent identifier:https://cran.r-project.org/web/packages/BayesTwin/index.html
Licence: GPLv2 or later
Publisher: Inga Schwabe
Version published: 1.0
Date published: 06/26/2017
Code repository
Name: GitHub
Persistent identifier:https://github.com/ingaschwabe/BayesTwin
Licence: GPLv2 or later
Date published: 11/09/2014
Language
R and JAGS
(3) Reuse potential
The BayesTwin package is accompanied by extensive documentation of functionality. Each function is accompanied by an R help file, which can be accessed by the user by typing help(…) with the function name inside the brackets. Each help file contains worked examples of real R code that users can paste into the R console and run. Furthermore, code examples are provided at the personal website of the package author. These also include code example on how to run the R package on a computer cluster.
Full open access is provided to all source code and full reuse rights via the generous GPL-Clause license. This makes it easy for others to use the code base, also in order to collaborate or ask questions.
A drawback of the method is that it is computationally intensive and can take several hours to complete. Future research therefore will focus on optimizing the MCMC algorithm procedure. Furthermore, individual twins in the data set with partly missing covariate data cannot be included in the analysis, leading to reduced statistical power. Another problem is that covariates cannot always be defined as a twin pair covariate or an individual covariate: some covariates can be shared for some of the twin pairs, but non-shared for other pairs. In future releases of the package, a new Bayesian method will be applied that incorporates covariates that can be both shared and non-shared, and that are given a prior distribution so that even individuals with partly missing data on the covariates can be used in the analysis [].